
Should we stop trialing relaxin drugs in pulmonary hypertension?
Tectonic's APEX trial, AstraZeneca's Re-PHIRE trial, and Merck's CADENCE trial

AstraZeneca’s phase 2 Re-PHIRE results in PH-HF
AstraZeneca ($AZN) tried a relaxin-mimetic (AZD3427) in patients with pulmonary hypertension associated with heart failure (PH-HF) in the phase 2b Re-PHIRE study. 220 patients were randomized 1:1:1:1 to receive AZD3427 1.1 mg, 5.4 mg, 30 mg, or placebo via subcutaneous injection every 2 weeks for 24 weeks. Main outcome was change from baseline to week 24 in pulmonary vascular resistance (PVR) as measured by right heart catheterization (RHC). They enrolled HF patients with NYHA II-IV with PH diagnosed by RHC (PAWP 15+ and mPAP 20+).
This is a broad group of patients! The patients had HF with any ejection fraction and isolated post-capillary PH (ipcPH) or combined pre and post capillary PH (cpcPH). This matters because cpcPH, defined as PVR > 2, has much worse outcomes than ipcPH. Also, the type of heart failure determines how the PH occurs and therefore the expected benefit of a given intervention. The destination is grossly the same (i.e., congestion of pulmonary veins and vascular remodeling) but the journey is different. For example, we would not routinely give ARNI to a patient with HFpEF but we would if the patient had HFrEF.
The AZN Re-PHIRE data presented at ESC HF 2026 was a miss [1, 2]. But, here’s what I’m seeing:
No safety signals
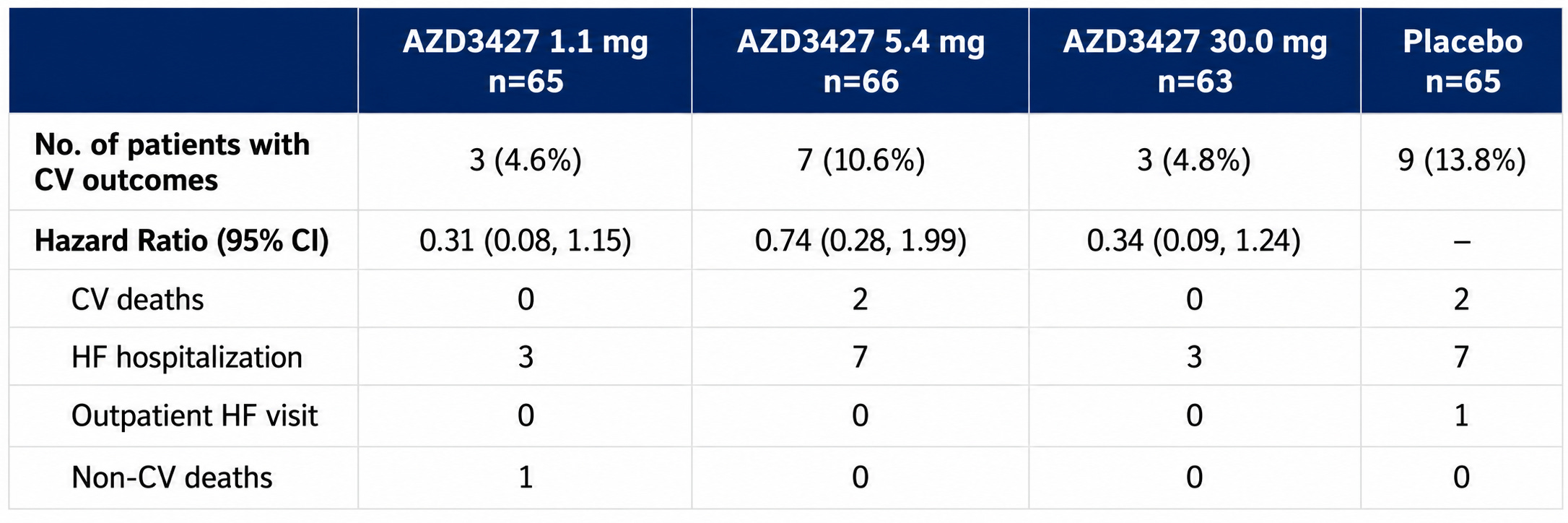
No real dose-response relationship. Admittedly, a bit weird. (Are we saturating the receptors? Or is the drug inert? Or a surprise third option?)
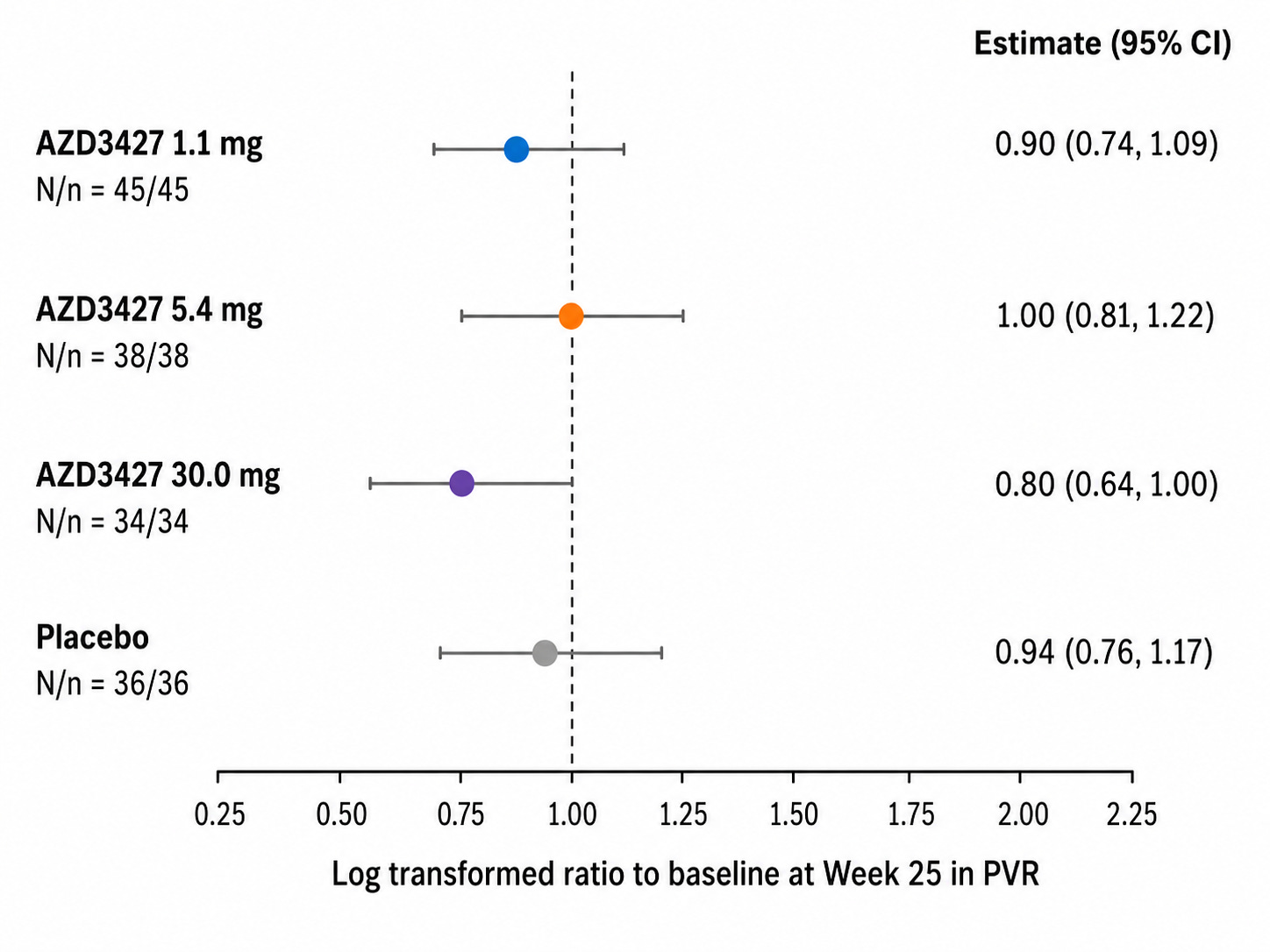
In PVR > 2 cpcPH-HF, here’s what the PVR change from baseline to week 25 looked like:
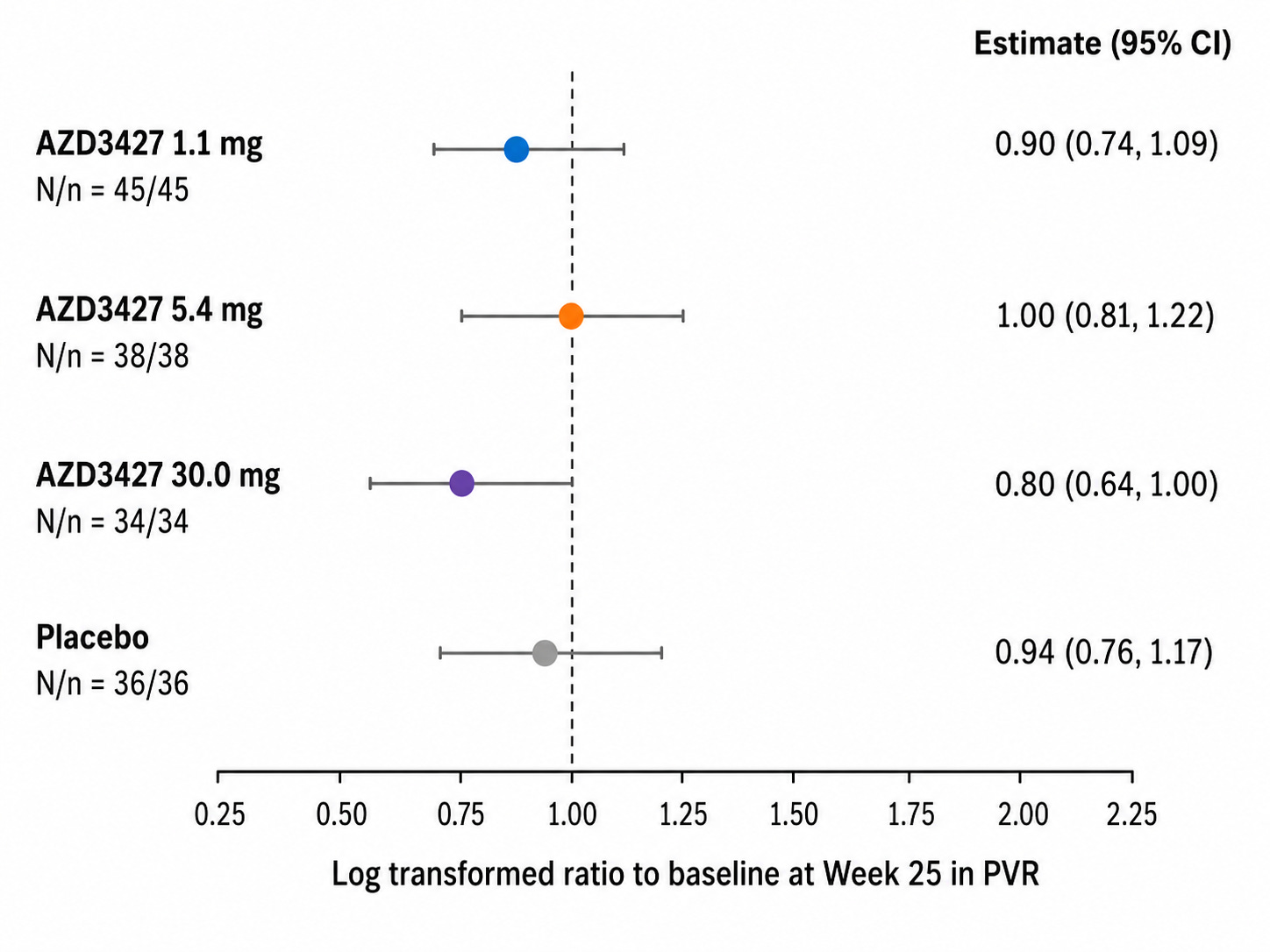
Remember that the cpcPH group shown above is HF with any EF
mPAP did not move in PH-HFpEF with treatment but at highest dose level (30 mg) PVR 95% CI did not touch 1. PVR = (mPAP - PAWP) / CO and though we can’t deconvolute patient level pre-post mPAP, PAWP, or CO changes, we can use the patient-aggregated values to make a vague inference. This suggests that CO increases with treatment in PH-HFpEF without safety signals (i.e., volume overload and clinical worsening). See below for time to clinical worsening curves:
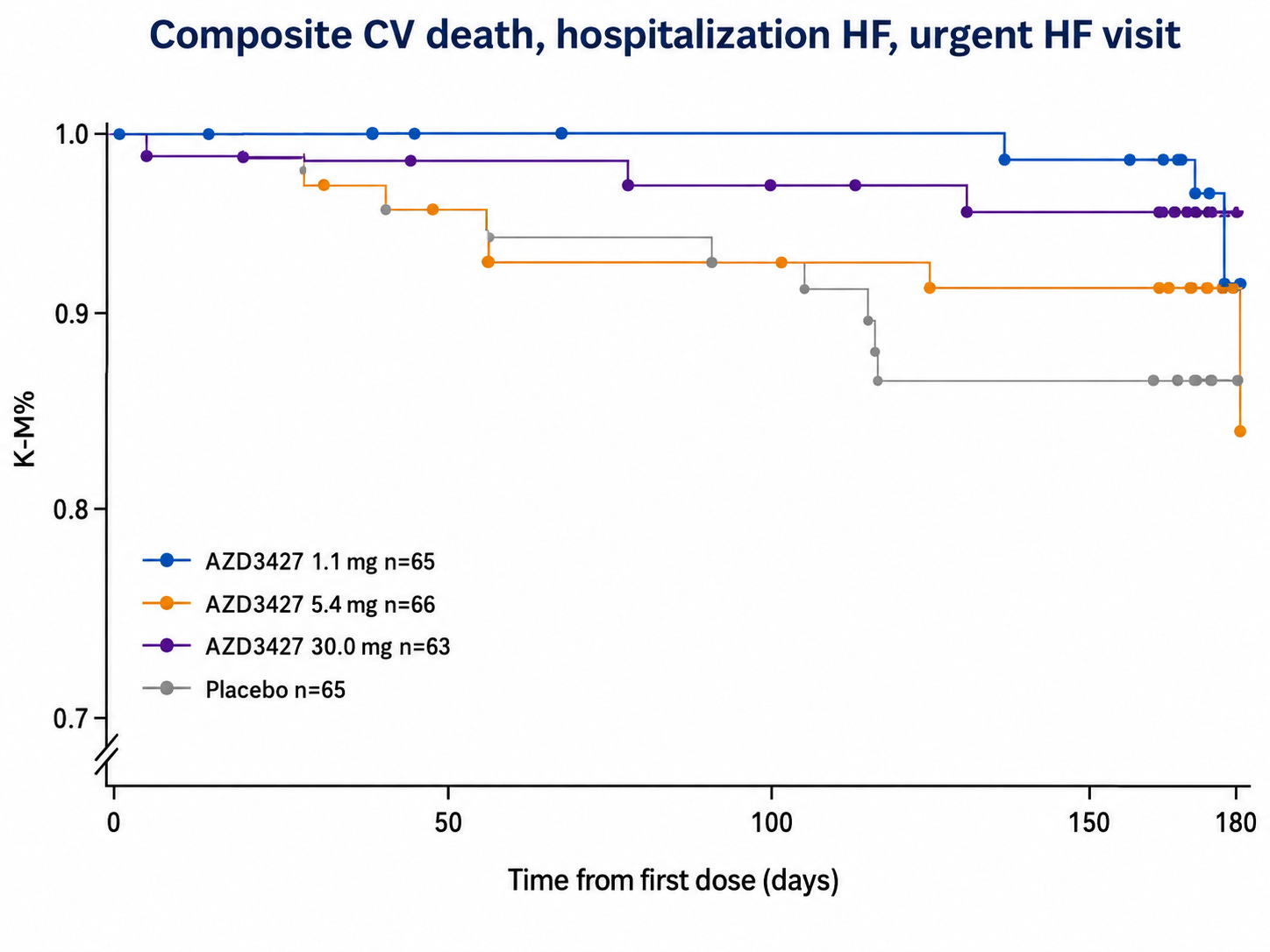
With the Re-PHIRE time to worsening curve in mind, we can see that the placebo time to worsening in Merck’s Phase 2 CADENCE trial of sotaracept in cpcPH-HFpEF (where median PVR was 5 and enrollment criteria was PVR 4+) was markedly worse than what we observe in Re-PHIRE.
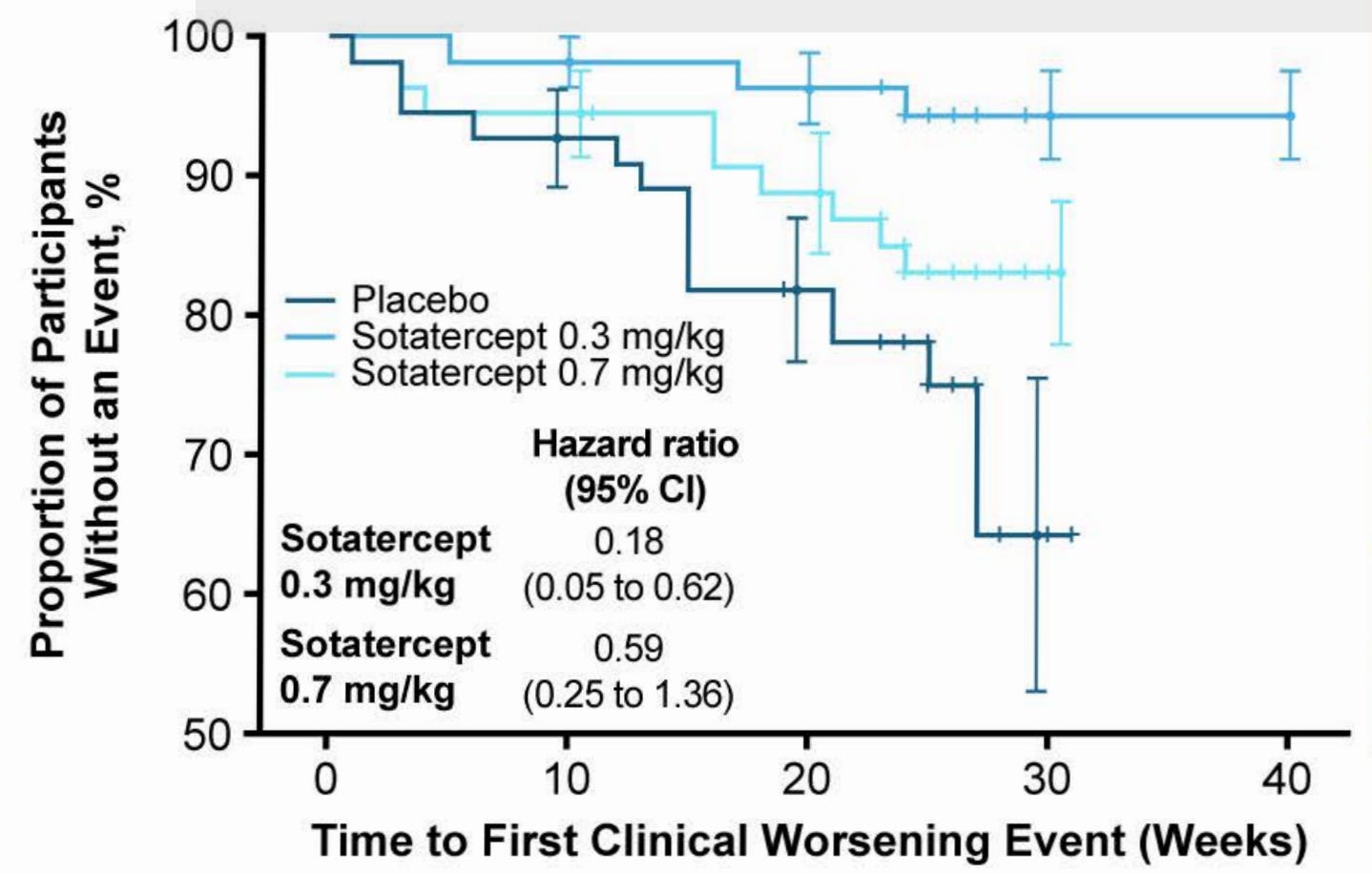
In CADENCE, 6% and 16% in 0.3 and 0.7 arm, respectively, had a clinical worsening event compared to 26% of placebo. Placebo in RE-PHIRE only had a 14% event rate. A combination of three things may have occurred: a) better background medication management in Re-PHIRE trial, b) cpcPH-HFpEF (PVR 4+) is a severe subgroup of PH-HFpEF disease, c) Re-PHIRE followed to ~25 weeks whereas CADENCE followed to ~30 in the placebo arm and we observe a number of events between week 25-30 in CADENCE placebo. This discussion of placebo arms implies that Tectonic’s APEX study may have a placebo arm like that of CADENCE out to 25 weeks. The Merck trial population is basically the same as what APEX is enrolling. Therefore, I would expect placebo to perform the same in both trials.
I would also like to note that CADENCE 6MWD was inconsistent across dose levels and actually marginally worse than placebo at the highest dose. I understand this was an exploratory endpoint in this trial, but it’s still worth the commentary.
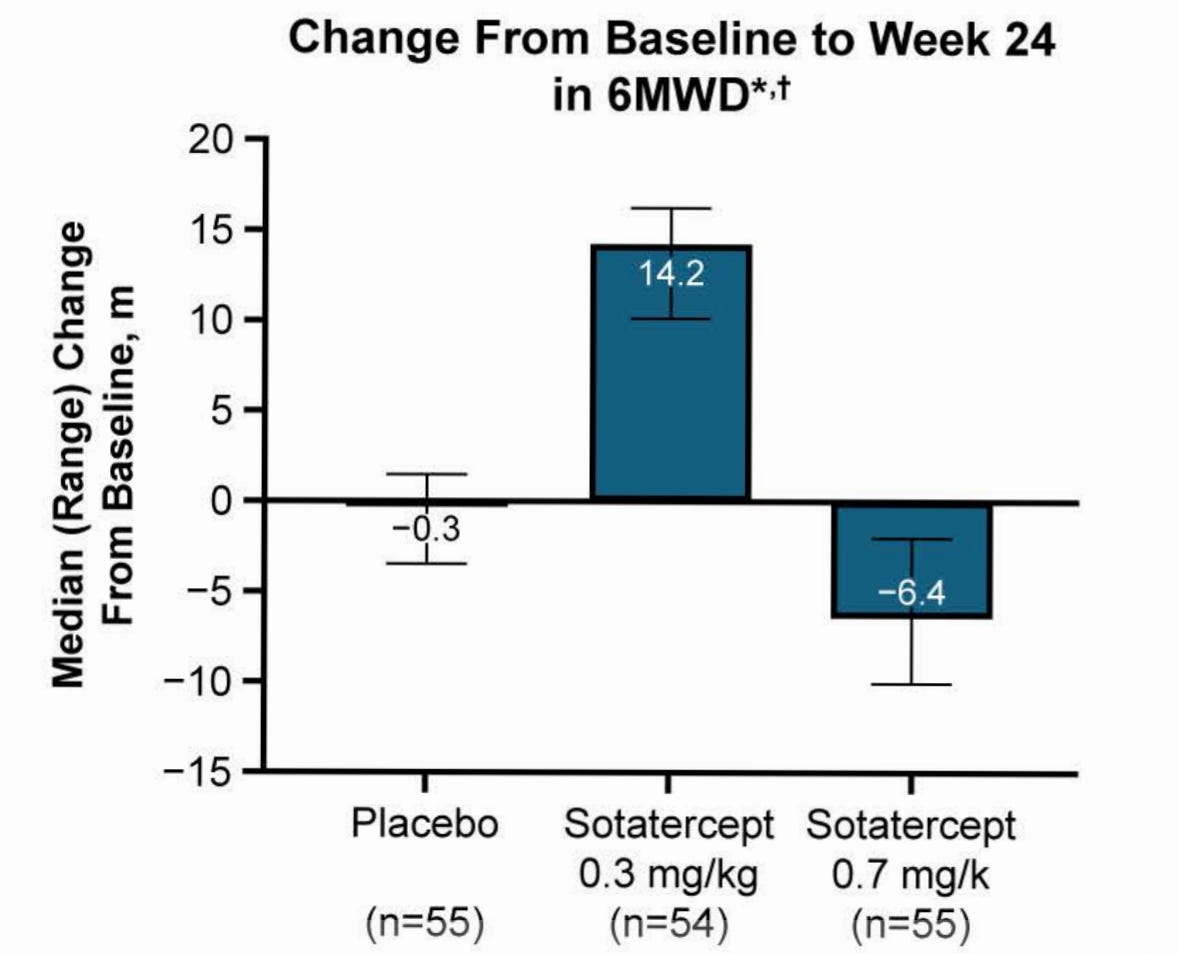
6MWD is noisy and tough to show improvement. This is the bar to clear, and it’s not a very high bar. Dire unmet need.
Tectonic Therapeutics’ Phase 2 trial of TX45 in cpcPH-HFpEF
I have posted previously (1, 2, 3) about why I think Tectonic has a differentiated offering than AstraZeneca when it comes to relaxin for PH for heart failure. In short, they have a better designed trial: one that looks at the population best suited to respond to relaxin in a positive way across the key parameters we care about.
Each relaxin drug program has learned from the ones prior to it. For example, Lilly’s relaxin trial with volenrelaxin enrolled patients with recent hospitalization for acute decompensated heart failure and from that trial emerged safety signals along with no effect. Since then, we have learned to not enroll recently hospitalized patients and enroll stable outpatients.
We used to set the cutoff for cpcPH at a PVR of 3, but have since relaxed this threshold. Based on cohort studies that have looked at PVR > 3 and PVR > 2, the main takeaway is that PVR > 3 have worse outcomes than PVR > 2. The principle here is that higher baseline PVR correlates with worse prognosis and simultaneously the place where intervention could produce the greatest effect.
Tectonic’s APEX Phase 2 trial is trying TX45, their relaxin mimetic, in PH-HFpEF NYHA II-III patients with 6MWD between 100-450 meters. Key group is cpcPH-HFpEF with PVR > 3. 180 patients will be randomized 1:1:1 and treated for 24 weeks. Tectonic will have 42 patients per arm with PVR > 3 cpcPH-HFpEF, numbers that parallel CADENCE sample size. Change from baseline PVR and the trend in time to clinical worsening are the two main things to look at here in my opinion. Safety is a co-primary outcome on their clinical trials webpage but this is unlikely to be an issue, especially since AZN did not have even a touch of safety concerns.
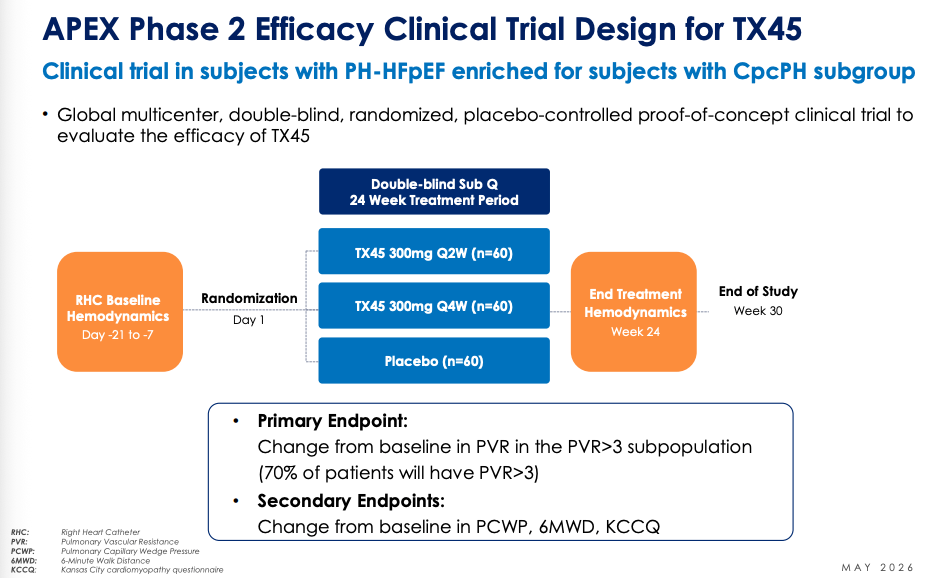
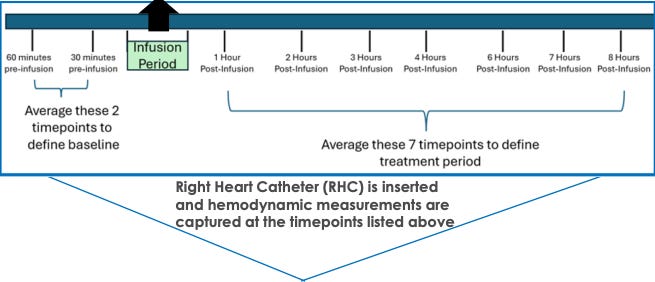
What might happen to PVR at 24 weeks? We have Phase 1 data where TX45 was given IV and RHC measurements were taken in an 8 hour window after infusion to determine effect on hemodynamics.
PVR change from baseline (CFB) 95% CI did not touch zero for the PH-HFpEF patients with baseline PVR 2+ and PVR 3+ with greater change in PVR 3+ group. The same story was not observed with HFrEF, the PVR CFB 95% CI crossed 0 for both PVR 2+ and PVR 3+.
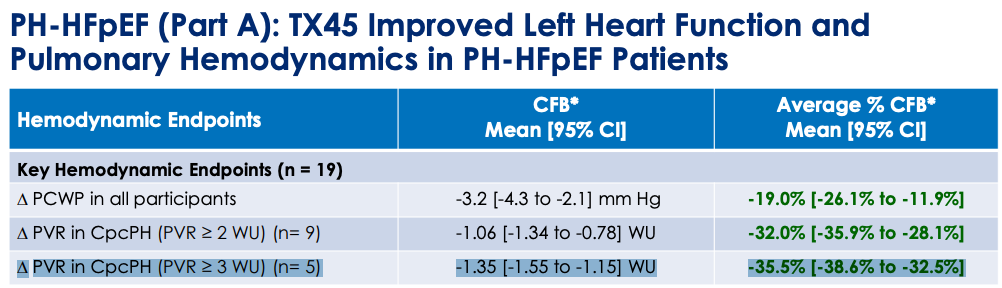
This is all the human data we have to go on, which raises two important questions for APEX: a) does the drug stick around for 2-4 weeks at a time or is it in and out in a day or two?, b) were these hemodynamic CFBs not due to drug but some other cause? For the former question, I leave that as an exercise for the reader to decide after reading the PK/PD tea leaves. For the latter, we must ask whether background medications or simply the process of being observed, lying in a hospital bed for ~10 hours, and having a RHC done 10 times could create some or most of the CFB we see here. However, the null effect in this scenario is not a likely cause for the hemodynamic changes: 1) hemodynamics are harder to influence than other surrogates (e.g., symptom severity surveys) simply by instrumentation, 2) background medications would have to be administered between hour 1-8 post-infusion and since the average across the 8 RHC’s are used, it would have to be done early in the window to move the average. Continuing on this idea more, I find it unlikely that the trial protocol allowed for diuretics to be given outside of clear signs of volume overload or that any other medication that could impact volume status or BP, for example, were allowed in the 8 hour window unless absolutely necessary. The limitation we can’t get around is that the TX45 cpcPH-HFpEF (PVR > 3) is based on data from five patients, so with larger sample size maybe everything goes to zero effect.
We shall see what happens with APEX. Topline results are expected late Q4 2026 or early Q1 2027.
Acknowledgements
Thanks to Adu Subramanian for discussions and back and forth; opinions here do not necessarily reflect his thoughts.