
DNA Foundation Models and Their Applications
What can we do with DNA Foundation Models?

Technical areas for improvement
(Data, data, data + building a reinforcement learning environment for biology)
State-specific promoters (CAR-T, AAV gene therapy, and ddRNAi drugs)
Discover new disease-causing targets through in silico mutagenesis
This post is already going to be really long. So, I will publish a Part 2 soon covering four more applications: biomarkers / patient selection strategies, DNA models as a service, predicting the cell-level effect of multiple genomic changes, and optimizing crop genomes and drug manufacturing systems.
Introduction
DNA Foundation Models (FMs) are large language models capable of 1) generating DNA sequences at scale that capture the grammar of DNA and 2) predicting diverse genomic properties (e.g., pathogenic variants) with minimal additional data or modeling. Before we jump in, let me tell you why you should care about DNA models.
Protein models like ESM3, AlphaFold3, Boltz2, and RFDiffusion generate, characterize, and design proteins thereby allowing us to make new drugs and discoveries. However, proteins compose just 2% of the human genome. What can we learn from the other 98% of the genome? If we can generate, predict, and characterize DNA sequences, what can we create?
Protein models primarily train on the protein data bank, a set of ~200,000 protein structures. In comparison, 490,000 whole genome sequences were deposited in the UK Biobank on August 6, 2025 alone. We have orders of magnitude more DNA sequencing data than protein sequence/structure data. The data advantage will grow even wider given that the cost of DNA sequencing and other molecular assays continues to drop. DNA FMs allow us to ingest this data and learn complex patterns.
This post is, in some ways, a response to Owl Posting’s ‘so-who-cares’ take on DNA models. DNA models have a lot of potential and it is early days both on the model building and application-side. Long story short, DNA models can be useful if coupled with high-quality domain-specific multi-modal data.
Survey of DNA Models
Arc Institute’s Evo2 (Feb 2025) and DeepMind’s AlphaGenome (June 2025) are the frontier models. Both models are open source.
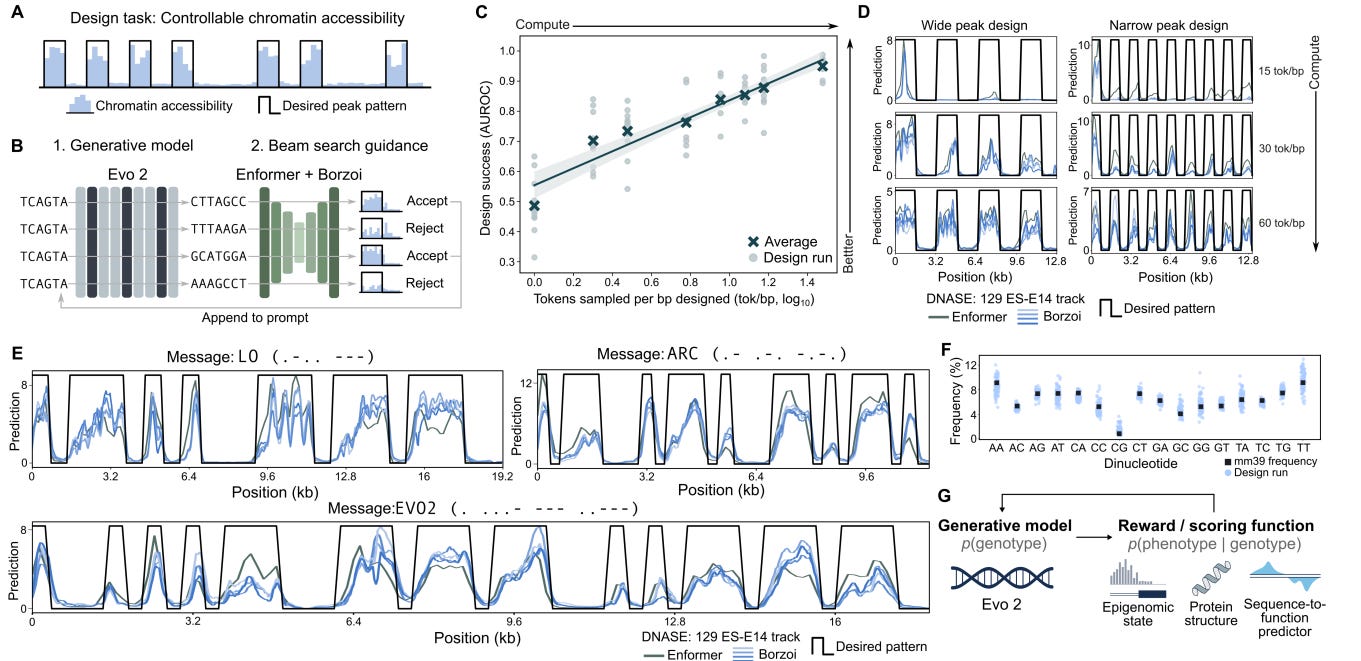
Evo2 is a 40B parameter 1M nucleotide context model that uses the StripedHyena2 (convolution and attention layers interwoven) architecture with nucleotide-level tokenization. Evo2 is trained on 9T DNA base pairs across all domains of life. Evo2 is able to generate sequences with specific epigenetic properties via inference-time search. Specifically, Evo2 was coupled with a guidance model (a prediction model that spits out a score given an input) to iterate and optimize the sequence to have certain properties (see figure below). Important caveat: we do not have experimental validation that the generated sequence actually creates the predicted chromatin accessibility.
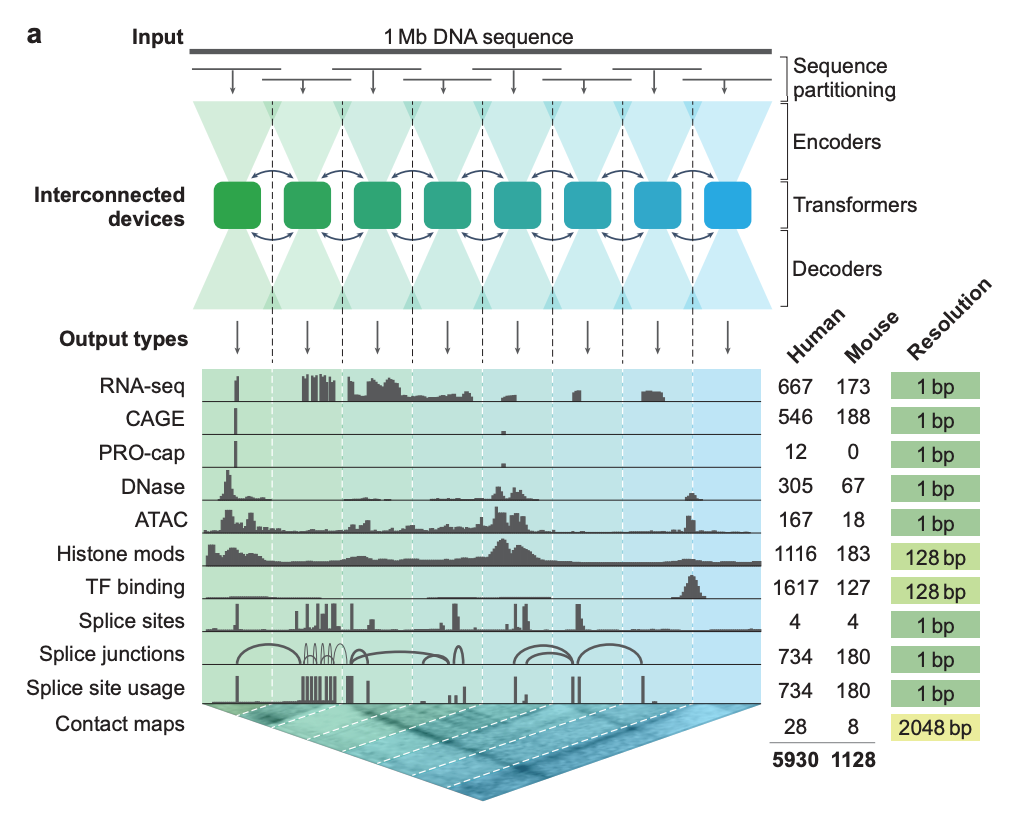
AlphaGenome is a 450M parameter 1M nucleotide context model that uses an encoder-decoder architecture composed of convolution and transformer structures with single nucleotide-tokenization. It was trained on multimodal sequence data including RNA-seq, DNA sequences, and Hi-C genomic contact maps (see figure below). AlphaGenome predicts many genomic tracks across cell types in human and mice and is competitive or better than top task-specific models.
There are several other DNA FMs including JanusDNA (Fudan University), Caduceus (Cornell), NucleotideTransformer (InstaDeep), GENErator (Alibaba), DNA-BERT2 (Northwestern), and LucaOne (Alibaba). They make different modeling and data choices including k-mer tokenization, shorter context windows, different neural network architectures, and smaller training datasets with limited genetic diversity.
But, how do we generate DNA sequences from a DNA model? Three key ways. Autoregressive sampling (GPT-style next token prediction), diffusion sampling, or Dirichlet flow matching (DFM). DFM is a smooth diffusion process that allows a guidance model to steer all positions in the sequence. Autoregressive sampling is the standard (used by Evo2) though Dirichlet flow matching holds significant promise for generating sequences subject to constraints. DFM is faster and enforces global constraints but can be complex to train and less intuitive to grok than autoregressive sampling. We can work through these negatives to realize the advantages of DFMs. Dirichlet flow models are a natural choice for achieving guided sequence generation.
Technical areas for improvement
This is mostly a section of me asking questions and speculating. Feel free to skip, not super relevant to the rest of the post.
1. Data:
The quality of training data strongly influences the type of problems the model can solve, its performance on those tasks, and its ability to generalize. Open questions include: How much raw data can we acquire? What data should we collect to maximize the model’s ability to generalize to unseen states?
What other modalities of data should we acquire? What additional data should we collect to fine-tune a general model for our specific set of tasks? In short, it depends on the application. But in general, we can collect high-quality data for fine-tuning models by developing scalable assays that directly measure the molecular properties of interest. One example is the recently released Variant-EFFECTS assay developed by Eric Lander, Anshul Kundaje, and Jesse Engreitz, which quantifies how each bit of regulatory DNA influences gene expression. Other labs and companies have large amounts of data relevant to training DNA FMs including Octant Bio, Jay Shendure, and Pardis Sabeti.
2. Model building:
How do we capture long-range effects without forgetting small-scale patterns? One option is to scale context length. The context length of 1M base pairs is the longest range current DNA FMs can handle. If we can increase context length 1.7 orders of magnitude to 50M, the length of the smallest human chromosome, we can capture chromosome-scale interactions. They are rare but their effect sizes are often large (e.g., BCR-ABL1 fusions in leukemia). But, with increased context length comes memory, compute, training cost increases and the risk that the model will forget small-scale features. What new types of architectures allow for ultra-long-range pattern recognition while preserving short-range capabilities subject to cost, compute, and memory constraints? This is an open problem. For example, Evo2’s StripedHyena2 architecture offered a solution to this problem by using interwoven convolution and attention layers.
Caduceus, a DNA FM, implements reverse complement (RC) equivariance which models the symmetry of DNA’s structure and base pairing rules. It also implements bidirectional learning such that it ingests sequences left-to-right and right-to-left to extract patterns. Could RC-equivariance and bidirectional learning improve Evo2’s performance? What other biologically-informed architectures should we try to implement? These are open questions.
How can we map DNA FM embeddings to biological processes? Sparse autoencoders (SAEs) are one option. Interpretable biological signals using SAEs have been explored for protein models (ReticularAI), DNA FMs (Goodfire) and sc-RNAseq data (Markov Bio) with varying degrees of success. Some other interpretability methods for biological deep learning models include DeepLift and TF-MoDiSco.
3. Optimization environment:
A generative model coupled with a prediction model can iterate to create arbitrarily large DNA sequences with a specific set of functions if the set of functions can be translated into a score that can be optimized. Think Prime Intellect for reinforcement learning in biology.
4. Benchmarking:
Lastly, we need benchmarks to score DNA FMs without significant test-train leak. There are a few datasets people use including TraitGym, CAGI, Genomic Benchmarks, and BEND but each DNA FM paper uses a different set of datasets to measure performance. The field needs a consensus benchmark. Relatedly, there is no METR equivalent cataloging model performance in this space. There probably should be.
Applications
What can we do with DNA models?
A. Designing state-specific promoters for gene therapy and biological research
Regulatory DNA elements (ITR sequence, polyadenylation signal, promoter) and a therapeutic transgene packaged in Adeno-associated virus (AAV) capsids are a type of gene therapy approved to treat genetic diseases including retinal dystrophy (Spark Therapeutics), spinal muscular atrophy (Novartis), and Duchenne muscular dystrophy (Sarepta). Modifications of the each of the three regulatory elements in the AAV impair viability of the transgene in human tissue and alter transgene expression. Also, the DNA in the AAV capsid is limited to ~4700 bases due to space constraints in the capsid.
We currently design regulatory elements from rules (e.g., OnTarget), experimentally characterizing the search space of regulatory elements, or bioinformatically scanning databases hunting for a target tissue advantage. Can we engineer regulatory elements that allow the transgene to be expressed only in target tissue? Yes. We can leverage DNA FMs to create tissue-specific regulatory elements. AlphaGenome predicts cell-specific chromatin accessibility (ATAC-seq, DNAse-seq), promoter marks (histone modidifcations), and CAGE (transcription initiation). We can design an objective function based on a composite of these measures being activated in the target tissue and not activated in off-target tissue. Then, we can generate sequences perhaps through Evo2 or DFM and implement our guided generation to discover and rank candidate promoter sequences that are specific to a certain tissue type.
Efficiently designing tissue-specific promoter sequences enables AAV gene therapy, CAR-T therapy, DNA-nanoparticle gene therapy, ddRNAi drugs, and basic science work (lineage tracing, calcium physiology, cell ablation, tissue-specific disease models).
CAR-T promoter design matters a lot. It impacts anti-tumor effectiveness and side effects. Typically, always-on promoters are used to create predictable anti-tumor activity. But, always-on promoters can create significant side effects (i.e., cytokine release storm) when administered in the short term. In the long-term it can lead to T cell exhaustion which means that signaling gets shut down and cannot work to kill cancer cells anymore. We can design T cell-specific promoters with activity levels that are optimized to prevent exhaustion and severe cytokine release storm with DNA FMs. Typically, CAR-T cells are edited outside the patient’s body but recent efforts have shown that we can create CAR-T cells inside a patient’s body by giving a patient a lipid nanoparticle containing mRNA. The promoter design question becomes even more important for in vivo CAR-T therapies developed by companies like Capstan.
Looking forward, designing state-specific regulatory elements may be a new avenue for therapeutics. Instead of maximizing expression for liver tissue, we can maximize expression for hepatocellular carcinoma cells or hepatocytes with dysfunctional glucose regulation (i.e., Warburg effect).
There are several groups pursuing ML-based strategies for regulatory element design. Ryan Tewhey, Chris Reilly, and Pardis Sabeti published their deep learning model for designing tissue-specific regulatory elements across three cell lines in Nature (Oct 2024). Ginkgo Promoter-0 technology predicts promoter activity in cell types using Borzoi. Ginkgo gene therapy solutions offers AI-driven sequence optimization of cell-type specific promoters within 3 months (too long in my opinion) and licensing of their promoters. AskBio designs synthetic tissue-specific promoters using their own bioinformatics engine. Meira identifies tissue-specific mini-promoters using high-throughput experimental assays.
B. Discovering novel disease-causing targets through in silico mutagenesis
DNA FMs are a general platform that can identify disease causing variants. A clear example of the potential for DNA FMs to achieve this is demonstrated in a study published by DeepGenomics in 2020.
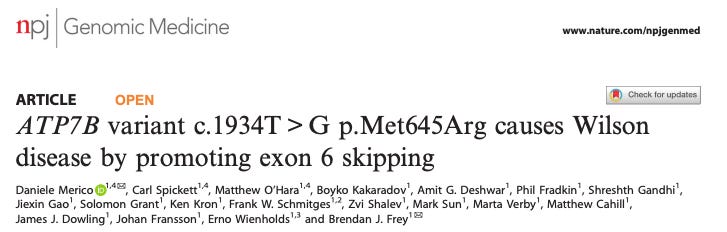
They used a deep learning DNA model to accurately describe the role of a missense mutation in Wilson’s disease. It was known that the mutation was associated with the disease but its mechanism was unclear given it had no effect on protein function in vitro. This study revealed that the mutation results in exon 6 skipping which enables the development of a new treatment for Wilson’s disease patients with this variant: steric blocking antisense oligonucleotides to restore splicing.
Also, DNA FMs can categorize missense mutations in proteins labelled as likely benign by AlphaMissense.
AlphaMissense classified 105139 mutations as likely benign. In ClinVar, 5108 of these were actually not benign. AUPRC is 0.18 for AlphaGenome on this very tough dataset. The prevalence of pathogenic variants is 4.8%. This is the expected performance if we randomly guess labels in this data. AlphaGenome does a lot better. I think the low AUPRC may not catch some people's eyes but I think its a great result. It shows an area (of many) where DNA models perform better than their protein counterpart
DNA FMs may have an advantage over protein models in predicting pathogenicity status of protein coding variants. With a multi-modal DNA FM, one can probe the effect a specific variant has across cell types and genomic tracks (e.g., expression, chromatin state, etc.).
Experimentally, scientists can make a specific change to a cell’s genome, observe its effect via molecular assays across cell types, compare their effect sizes and rank-order them by disease causing potential for further investigation in mice or other organisms. Issues with this approach include the cost and time of systematically probing the 3B base pair human genome and investigating the effect that cell-level changes like expression or chromatin state have on an organism’s phenotype. Consulting the scientific literature can aid in disentangling the cellular effect to phenotype linkage but is often sparse and hard to interpret. However, if we can prioritize the changes we wish to investigate (even loosely), we can then employ DNA FMs to quickly characterize their cell-level changes in silico and select a small set of variants for subsequent experimentation and de-risking. AstraZeneca fine-tuned a DNA FM to predict noncoding variant pathogenicity, so this topic is clearly top-of-mind for big pharma. The disruption of regulatory genomic elements (e.g., splice sites, promoters, enhancer, transcription factor binding sites) are understudied and DNA FMs allow us to probe into the effects of regulatory elements.
The process for choosing the changes to investigate by DNA FMs can be informed by clinicogenomic data. Two specific avenues for doing so are rare disease WGS data and human genetics (e.g., GWAS) studies. If a patient has a set of variants discovered by WGS and an observed phenotype (e.g., lab values, clinical syndrome, imaging), then we can investigate in silico, without assembling specific datasets, which one (or set of them) may lead to the phenotype. Companies that 1) sequence diverse populations or 2) assemble rare disease datasets are best positioned to leverage this technology.
(1) Sequencing diverse populations: We hope to emulate the process that yielded PCKS9 inhibitors for cholesterol management. Essentially, naturally occurring PCSK9 variants were observed to be associated with lower LDL and cardiovascular events in select populations. Then, mechanistic work was pursued in cell and animal studies. For example, VariantBio, a company working to sequence diverse individuals, could uncover future drug targets. Their downstream work could be aided by DNA FMs.
(2) Rare diseases: The undiagnosed diseases network (UDN) aggregates rare disease clinicogenomic patient data and seek to discover the genomic determinants of their conditions. They recently introduced a statistical method for prioritizing disease genes and discovering new diseases associations based on pooling WGS data across rare disease patients, finding recurrent mutations in genes, and grouping across pathways. UDN also works to deploy model organisms to characterize variant-phenotype relationships. Arcadia Science also works to develop new model organism systems and may be positioned to leverage DNA FMs to quickly test variant-phenotype relationships. Citizen Health is a rare disease company using AI to define trial endpoints and identify patient clusters to run trials. They have longitudinal clinical data (and potentially genomic data) that may enable variant to phenotype predictions. In silico assessment of variant to phenotype relationships also accelearte N-of-1 gene therapy treatments too.
C. Resolving variants of uncertain significance (VUS)
FDA-approved targeted therapies require patients to be ruled-in for eligibility based on if the patient has a pathogenic mutation in a predefined set of genes. Companion diagnostics offer tests to determine molecular eligibility and rule-in patients if they have a known pathogenic or likely pathogenic mutation. However, many patients who receive testing have only VUS and are generally not eligible for therapy. Also, as more patients receive WGS, more VUS will be reported. VUS reflect our scientific uncertainty about whether the variant causes the gene product to become dysfunctional and contribute to disease phenotype. DNA FMs can investigate VUS in silico to characterize their deleterious effects. What evidence do we have that this might be possible? We can look at the Evo2 and AlphaGenome papers for a base case.
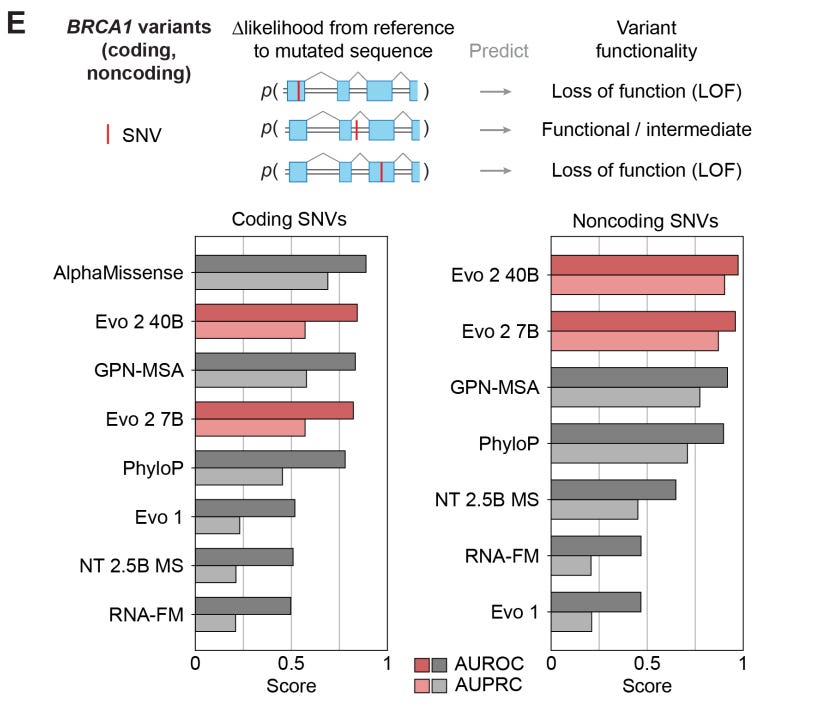
Evo2 was able to accurately classify BRCA1 coding and noncoding SNVs as pathogenic/likely pathogenic or benign/likely benign but VUS were removed. PhyloP, a much simpler algorithm, performs nearly as well, which has contributed to skepticism about DNA FMs for variant classification.
AlphaGenome examined the effect of clinically observed non-coding variants near TAL1 and accurately predicted the mechanism of their functional impact in acute myeloid leukemia.
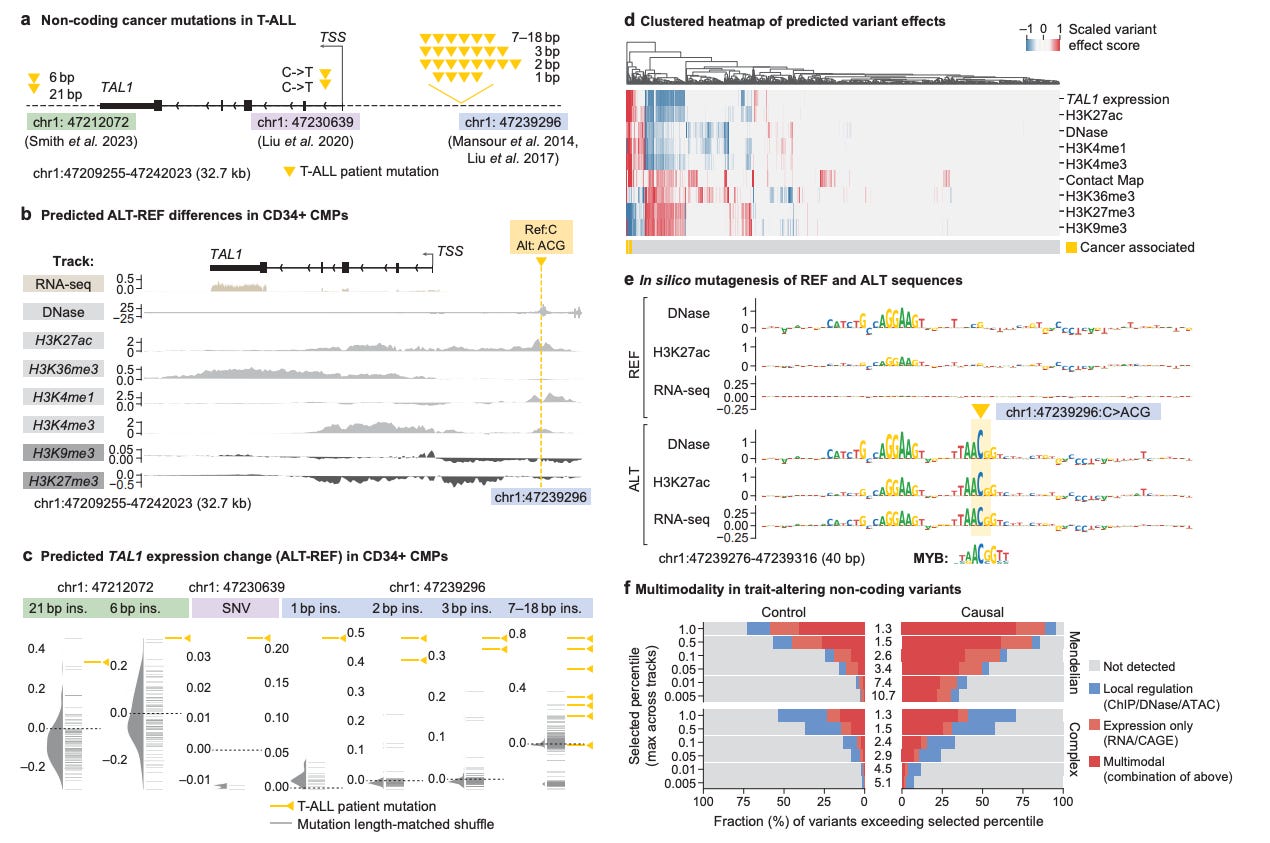
DNA FMs offer a computational way to bridge the variant-to-function gap.
Reclassifying VUS could materially impact sales of targeted therapies and health outcomes for cancer patients. In the US, HER2 negative breast cancer patients with germline pathogenic BRCA mutants are eligible for PARP inhibitors (PARPi). About 7% of patients receiving BRCA1/2 testing for breast cancer have VUS only. So, ~13,000 HER2- breast cancer patients have BRCA VUS each year whereas ~50,000 patients have BRCA pathogenic variants. Conservatively, if 5% of BRCA VUS patients are reclassified, 650 more patients are eligible. Assuming 180,000 new HER2- breast cancers in the US each year, $15k per month, 12 months of use on average, and 70% take-home after rebates, this translates to 81M per year in additional revenue if 5% are reclassified. PARPi is also indicated for prostate and ovarian cancers too. Reclassifying VUS at-scale could be a viable business model if value can be captured by the company doing the reclassification.
In this space, Velsera and QIAGEN QCI Intercept are software engines that serve labs and health systems to interpret variants. Myriad Genetics and Ambry Genetics develop clinical genetic tests and actively reclassify variants through RNA follow up, family studies, and surveillance. DNA FMs can reclassify at scale and therefore develop clinical tests that rule-in more patients for targeted therapy.
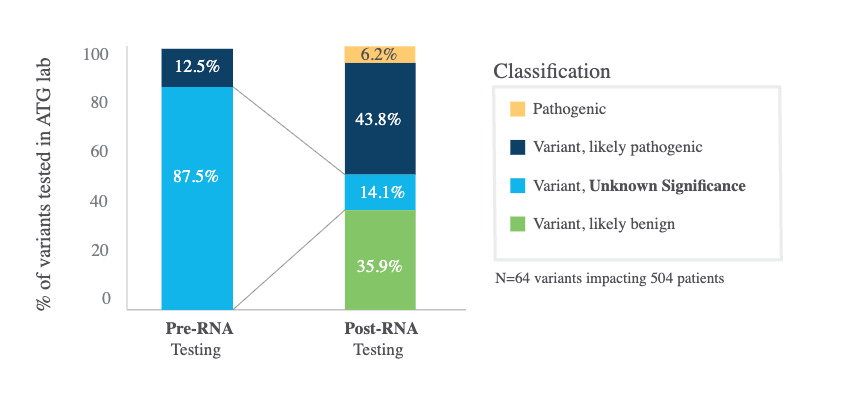
Myriad reported 25% of VUS were reclassified of which 9% were upgraded. Ambry reclassified a considerable number of VUS to likely pathogenic / pathogenic (see figure below, small sample size limits generalizability). These reclassifications are reported to ClinVar and then get used by other groups.
D. Biosecurity of generated DNA sequences
Given that bad faith actors can now generate novel DNA sequences with toxic or pathogenic features, we must develop technology to identify such sequences. In fact, Le Cong and Mengdi Wang wrote a paper showing how DNA language models can be used to generate problematic sequences. We need compliance software that can detect novel pathogenic sequences. DNA FMs are a solution to this problem.
Shelby Newsad and CompoundVC have written about the problem of DNA biosecurity:

I agree with their premise.
DNA synthesis companies (e.g., TWIST, IDT, Thermo Fisher, Ginkgo, Tierra, benchtop DNA printers) typically follow government guidance currently composed of rules-based sequence checkers and customer watchlists. This is not a good solution. We miss a lot of pathogenic sequences. A fine-tuned DNA FM can flag problematic sequences. Or DNA FMs can have enhanced safety alignments and tracing mechanisms. This is preferable over rules-based engines like UltraSEQ (TWIST is a customer) given that novel sequences with specific properties can now be generated at scale. Also, the current administration has expressed interest in using AI for biosecurity and we may be able to make progress on this front quickly at a national level.
I have a few more applications of DNA FMs that I will defer to part 2 of this series. If I could summarize this post in one brief sentence, it would be: DNA models can be useful if coupled with high-quality domain-specific multi-modal data.