
Semaglutide in early-stage Alzheimer's Disease
Looking ahead to the EVOKE and EVOKE+ Novo Nordisk trial readouts in Dec 2025

Novo Nordisk is presenting top line results from their EVOKE and EVOKE+ trials at CTAD on December 3 at 5:05 pm PST. EVOKE and EVOKE+ are two trials evaluating oral once-daily semaglutide against placebo in early symptomatic Alzheimer’s Disease (AD). Will semaglutide cure a patient with AD? No. Does it address the inciting pathological process and halt it? Also, no.1 But, will semaglutide help patients with early-stage AD function better both cognitively and do their daily acts of living? Yes. This post will attempt to convince you of this.
Novo’s US patent for semaglutide expires 2031 and as early in 2026 in China and Canada. Compounding pharmacies like Hims will continue to battle with Lilly and Novo. Novo market share continues to get eaten by Lilly both US and worldwide. Novo lost the Metsera acquisition to Pfizer. And their board got overhauled a few days ago. Novo appointed a new CEO in July of this year. This is why they trade at a 13.28 P/E ratio and sit near a 52 week low while Lilly rides high at P/E of 50 and near 52-week high. Novo management say this about their own trial in September: “We always presented it as a lottery ticket and it still is because it’s very uncertain” and their comments on their Q3 call echoed the sentiment. Morgan Stanley and other analysts “expects Novo Nordisk Alzheimer trial to fail”. Now that I have set the scene, we can talk science.
EVOKE and EVOKE+ are looking at semaglutide-placebo difference from baseline to week 104 in the clinical dementia rating sum of boxes score (CDR-SB). The CDR-SB survey captures cognitive and functional impairment across 6 areas: memory, orientation, judgement and problem solving, community affairs, home and hobbies, and personal care. Higher scores are worse. Improvements or slowing of decline on this axis is all that matters. It is irrelevant if the treatment treats the root cause, has a fancy mechanism of action, or improves some biomarker.
The trial population is 55-85 year old patients with mild cognitive impairment (MCI) or mild dementia due to AD with confirmed amyloid abnormalities on PET or CSF analysis. 104 week main treatment phase with 52 week extension. 8 week semaglutide dose escalation regimen (3 mg [0-4 wk], 7 mg[4-8 wk] and 14 mg [8-156 wk]). Plasma biomarkers and CSF will be analyzed too, these are exploratory. Up to 30% of overall trial population could have T2D, but no stratification by T2D status was conducted at randomization.2 Patients in either arm can be on standard of care (e.g., anti-amyloid antibody or memantine).
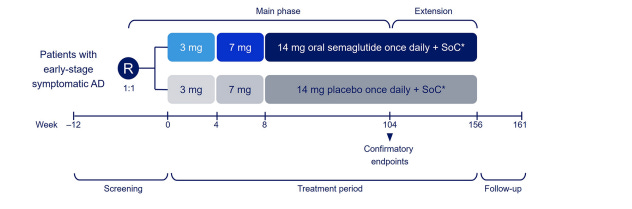
EVOKE+ allows patients with small vessel pathology (i.e., 1+ lacunar infarct plus/minus age-related white matter changes) to join. And we may find that small vessel associated dementia can be treated well by semaglutide, and that would be a broadening of their label beyond AD but for today, we will just think about early-stage AD patients. ECG, physical exam, neurological assessments, and markers (NfL, p-tau 181, GFAP, hs-CRP) at week 52, 104, 156. No futility or interim analyses are planned. In their exclusion criteria, they note patients cannot be on a GLP-1 in the 90 days prior to screening.
Why semaglutide will result in statistically significant improvements CDR-SB scores relative to placebo at week 104 compared to baseline?
Has there been an indication that semaglutide has NOT worked in? Diabetes, cardiovascular disease, obstructive sleep apnea, liver disease, and encouraging data in alcohol use disorder, opioid use disorder, and smoking cessation.
A 9-week, placebo-controlled study of 48 adults with alcohol use disorder demonstrated that once-weekly semaglutide reduced alcohol self-administration in the laboratory (β, −0.48 [95% CI, −0.85 to −0.11];P = .01), craving (β, −0.39 [95%CI, −0.73 to −0.06];P = .01), and drinks per drinking day (β, −0.41 [95% CI, −0.73 to −0.09]; P = .04) despite a short treatment duration in a non–treatment seeking population.
Primitive GLP1s have shown mixed results (3 positive, 2 negative) in Parkinson disease.3
Liraglutide, an older generation GLP1, led to successful results in AD. Phase 2b trial of liraglutide led to reduced shrinking in parts of the brain that control memory, learning, lanugage and decision making by nearly 50% compared to placebo and was presented at AAIC in 2024. However, a full paper was never released. But, Novo did fund the liraglutide study so you have to assume they understood what went on in the trial and made the informed decision to continue the semaglutide trials. Here is some more commentary from alzforum on the liraglutide trial:
According to the published trial protocol, clinical secondary outcome measures included change on the ADAS-Cog-Exec z score—which contains the ADAS-Cog and the executive function portion of the neuropsychological test battery—as well as the Clinical Dementia Rating Scale Sum of Boxes and the AD Cooperative Study—Activities of Daily Living (Femminella et al., 2019). Edison reported that on the ADAS-Cog-Exec, participants on liraglutide declined less precipitously over the 12-month trial relative to those on placebo, and that, at p<0.01, the effect was statistically significant. However, cognitive trajectories of the treatment and placebo groups largely overlapped, as did the error bars. Any difference between the groups was small, at best.
Edison reported that the drug curbed gray matter atrophy by 50 percent—a statistic then widely reported in the news. What was it based on? Participants in the placebo group lost a total of 13,500 voxels, or 1mm3 cubes, of cortical gray matter throughout the trial, while those taking liraglutide lost about half as many. To put those losses in perspective, both groups started the trial with some 555,500 voxels, meaning placebo and treatment groups lost roughly 2.4 and 1.2 percent of their cortical gray matter, respectively.
I agree that the imaging based primary endpoint reduction reported on here means nothing. Functional status is all that matters. With 80 patients in each arm, it’d be great if you could see a difference. But with 1800 patients in each arm (pooled across EVOKE and EVOKE+), the trajectories won’t overlap - we’ll know the answer for sure. I do think the p < 0.01 on ADAS-Cog-Exec was nice though, but I will chalk that up to statistical artifact to try to raise the bear case as best I can.
The liraglutide trial gave once-daily 1.8 mg subq injection of liraglutide for 48 weeks. EVOKE/EVOKE+ gives once daily 14 mg oral semaglutide for 104 weeks. We can look to PIONEER 4 to understand 15 mg PO daily semaglutide vs 1.8 mg SQ liraglutide. Weight loss at 52 weeks is grossly in favor of semaglutide, glucose control is in favor of semaglutide and both have similar discontiuation rate (8% semaglutide, 6% liraglutide) [Pratley et al 2019, Lancet]. 4 So in EVOKE/EVOKE+, we give a better drug for twice as long. Recipe for success.
Neuroinflammation is a plausible driver of AD.
Semaglutide may have limited ability to cross the blood brain barrier. However, studies in animals have shown that semaglutide interacts with the circumventricular regions and specifically accesses GLP-1R–positive brain regions following peripheral administration, including the area postrema in the hindbrain, the arcuate nucleus of the hypothalamus, and the lateral septal nucleus [50]. These observations suggest that semaglutide may have at least limited direct access to the brain as well as exerting peripheral effects
Semaglutide may be brain-penetrant at least to some degree. We do not fully understand how semaglutide works in the brain and perhaps its effects are neuroprotective through a mechanism we do not fully appreciate.
We do know that semaglutide curbs peripheral inflammation (hs-CRP, a marker of inflammation, decreases on semaglutide treatment). For example, in the STEP 1, 2, and 3 trials, semaglutide 2.4 mg weekly reduced hsCRP by 39–48% versus placebo at 68 weeks, regardless of baseline BMI, glycemic status, or CRP level [Verma et al 2022].
And peripheral inflammation can impact neuroinflammation. So, EVEN if you think semaglutide does nothing on the brain, it can exert anti-inflammatory effects on the brain by modulating inflammation in the rest of the body.
Although some benefits of GLP-1 medicines, such as improvements in obstructive sleep apnea and osteoarthritis, are largely driven by weight loss, other important effects appear independent of weight or glucose control. Large clinical trials such as SELECT (cardiovascular disease), ESSENCE (metabolic liver disease), and STRIDE (peripheral artery disease) have revealed weight-independent benefits, which may reflect reduced inflammation and direct effects of GLP-1 medicines on the heart, liver, and blood vessels.
It’s all glucose control. And weight loss. So why would it work in AD? This is the main point bears make. I agree with them that glucose control and weight loss do not stop AD from taking place or change the disease causing process itself. But, it could delay progression and offer quality of life improvements for patients with AD at least on par with the rather modest effects of anti-amyloid antibodies with significantly less cost, no risk of brain bleeds, and ease of use (oral pill versus IV infusion). I guess my point to the bears is, who cares if its all downstream of glucose control! The mean age was 72 years, ~15% had T2D, ADS-Cogs-13 was 27, mean MMSE was 24, and mean MOCA was 18.8 at baseline5. Common comorbidities in early-stage AD with these scores are diabetes, hypertension, and hyperlipidemia. About 50% of patients in this age group with around this level of cognitive scores have a BMI > 25. GLP1 is bound to help - we know it does. We also know semaglutide reduces MACE and offers improved glycemic control without the risk of hypoglycemia, so couldn’t everyone in this population benefit from the known effects of semaglutide?
Having more normal glucose levels improves cognition and slows disease progression in MCI. Microvascular dysfunction contributes to disease progression in AD. [Morris et al 2014, Wang et al 2022, Lin et al 2023, Gonzalez-Rellan and Drucker 2025]
Losing weight when BMI > 25 also improves energy and cognition [Verone et al 2017, Siervo et al 2011]
GLP1s improve BP [Krumholz et al 2024] which in turn can improve cognition or decrease the risk of disease progression [Reboussin et al 2025, Hughes et al 2020, Dallarie-Theroux et al 2021]
Semaglutide may do a lot of other cool things like regulate blood brain barrier integrity, promote homeostasis of microglia and astrocytes, positively effect the adaptive immune systemic, manipulate synaptic viability and offer neuroprotection. Ok, maybe6. I have no idea. I am going to assume that for the sake of argument it has a zero percent chance of doing any of this but it STILL makes sense that the trial will hit.
We have lots of observational data on AD and GLP1s. Observational data results are hypothesis generating and not proof of anything. But still, its better than having observational data with results saying GLP1s speed up AD progression or increase AD incidence. [Schechter et al 2025, Zhang et al 2025, Wu et al 2025, Tang et al 2025]
Semaglutide will slow disease progression of early-symptomatic AD at least to the level offered by anti-amyloid antibodies if not more solely on the basis of its ability to decrease weight, control BP, decrease peripheral inflammation, and control blood glucose levels. Controlling these variables will lead to improved cognition, as is already established. If semaglutide has other magical neuroprotective properties that is great but totally unnecessary to read out positive at CTAD.
Semaglutide is a tolerable medication with a ~8% discontinuation rate and no risk of brain bleeds or necessity for IV infusion (compared to anti-amyloid drugs for AD patients). Semaglutide is cheaper than any other option out there and will continue to get cheaper as more competition arises, patents run out, and government sets new policies.
I am not saying the trial will hit. I am saying the probability of success is greater than what the market thinks. To be fair, a positive result here would move Novo stock like 5-10% in my opinion so not much.
Clearing amyloid-beta plaques in the brain doesn’t seem to work well. They slow the rate of disease progression a little bit but not much. You can argue that those trials didn’t offer the amyloid clearing drugs early enough in the spectrum. Or that the risk of brain bleed is significant enough to make this not a useful enough therapy and that amyloid beta is the culprit but we just need a better way to clear it. I won’t argue with you. But, clearly, there is a large unmet need for patients with early-stage AD.
That’s odd, I would think they would want to make sure groups are balanced for T2D at the start to avoid questions about one trial have an overwhelmingly large amount of T2D patients biasing results. But, law of large numbers (~1800 patients) says the probablility of meaningful imbalance in T2D patient fractions in trial arms is slim.
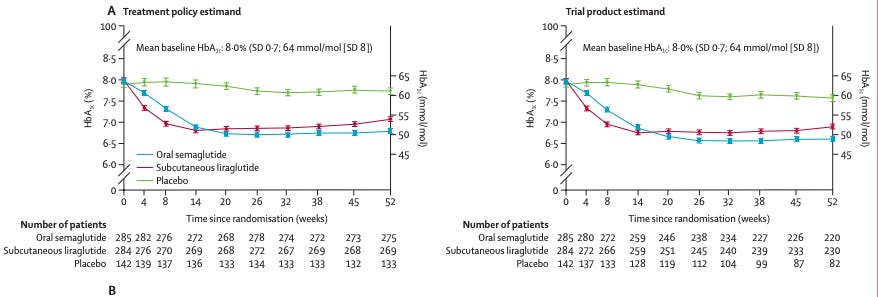
Charts of weight loss and A1C on semaglutide and liraglutide treatment.

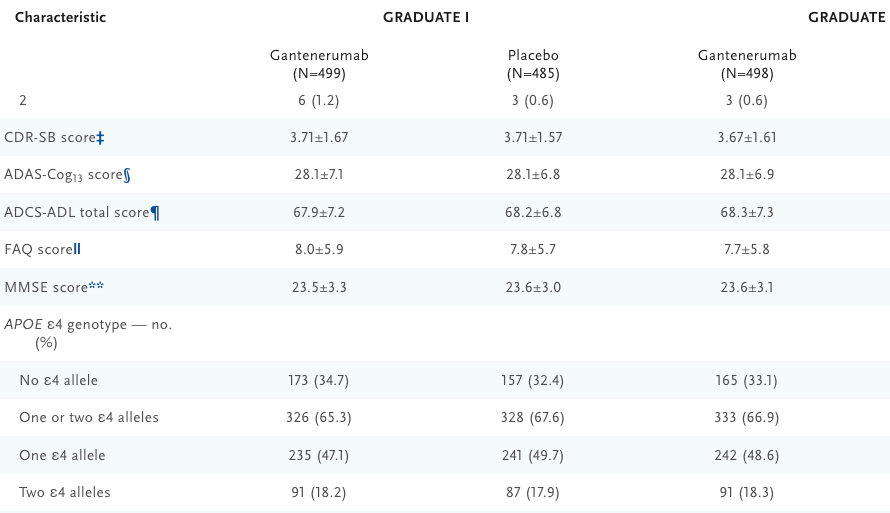
These characteristics are roughly what was observed in the anti-amyloid GRADUATE I and II trials (https://www.nejm.org/doi/full/10.1056/NEJMoa2304430) even down to the APOE e4 homozygous rate.
Please see references 17-26 here from the EVOKE and EVOKE+ paper

Didn't work
This piece really made me think. Your insightful analysis on pragmatic benefits of semaglutide for early AD is incredibily important. Thank you.