
Understanding the science behind the gene editing treatment for Baby KJ
A workflow for the rapid development of N-of-1 gene editing treatments

TL;DR: There are many other such inborn errors. Unfortunately, KJ’s story is not unique. For much of human history, we have had no option to help. But, now we can. Can we develop n-of-1 treatment for rare inborn errors of metabolism and give it to patients before they develop irreversible effects from their disease? We have the technology (base editors, prime editors, bridge recombinases, LNPs with targeting mechanisms like SORT LNPs and targeted LNPs), sequencing technology that keeps getting cheaper, and software. The targeted LNP engineering and tissue distribution/toxicity is arguably the most unsolved part of the science. This is now an engineering and clinical medicine problem. We can solve this. FDA is committed to helping address this issue. More or less this looks like: define clinical problem (sequencing, clinical input from MDs), design gene editing system (LNP, editor), assess edit efficacy + off target effects1, find dose and perform toxicology studies.
We are going to dive into the landmark NEJM paper by Kiran Musunuru and team. From this, we will understand the science behind base editing, what goes into making a successful gene editing product, think about platforming gene editing treatments, and consider open clinical and scientific questions in this space.
Baby KJ was born with carbamoyl phosphate synthetase 1 (CPS1) deficiency, a rare genetic disease. 1 in 1.3 million are affected with this disease and 50% of those affected die in early infancy. CPS1 deficiency (CPS1D) prevents the urea cycle from making urea and offloading ammonia, a waste product generated from breaking down proteins (see figure below). This process primarily occurs in the liver and causes hyperammonemic crises (temperature instability, respiratory distress, seizures, coma) and irreversible neurologic injury.

Patients with CPS1D often end up with irreversible organ damage or die before they grow large enough for liver transplantation. So, how did this team of scientists and clinicians treat KJ?
To treat something, you first have to recognize that it exists. They noticed KJ had an ammonia level > 1000 μmol/L (normal in newborn is 37-118 μmol) which suggested that KJ had a urea cycle problem. Blood profiling of amino acids suggested a proximal urea cycle defect. Subsequent targeted sequencing identified two variants C10003T and G2140T in the CPS1 gene that caused a Q335X and E714X mutation, respectively. X signifies a stop signal that prevents further translation of the RNA sequence. The C to T mutation occurred on the paternal allele and the G to T occurred on the maternal allele, knocking out both copies of the CPS1 enzyme. The key to detection of KJ’s condition was the pediatricians flagging a high ammonia and then performing targeted sequencing. Only a few states (Florida, Maine, Massachusetts, Mississippi, New Hampshire, Rhode Island and Vermont) include CPS1 in the newborn screening panels. Even still, these screens cannot reliably identify all cases of this disorder. If we sequence (with enough resolution) each child and can turn around results in less than a day, we can catch many such rare genetic diseases. In a 2023 JAMA pediatrics study, of the 297 variants found on either WGS or targeted sequenced in hospitalized infants with a suspected genetic disease, 201 of them were due to unique single nucleotide substitutions. Assessing clinical significance between mutation and phenotype can be challenging when the connection is not as straight forward and validated like CPS1D. This remains an open problem2. However, this establishes the precedent that the majority of genetic diseases, if identified, can be treated with the exact technology set forth in this paper.
From sequencing and biochemical labs, the team realized that KJ had effectively zero CPS1 activity. Based on mouse models, we know that if CPS1 activity is back to at least 10% of normal we can restore plasma ammonia and amino acid levels to within normal limits. Clinical observations in humans confirms that partial activity around 10% is sufficient. But, where does urea cycle activity take place? Which region of the body do we need to target? Almost all urea cycle activity happens in hepatocytes, primarily in periportal hepatocytes (region 1 in below figure) but also in perivenous hepatocytes (region 3 in below figure).
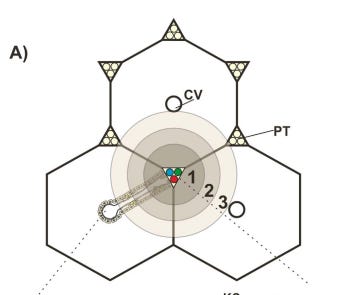
Blood from the branches of the portal vein and hepatic artery flows as a mixture through the sinusoids to the central vein. During this process, metabolism and re-composition of blood occurs (see figure below for high-level breakdown of processes including urea cycle activity). Our edit system ideally must selectively reach hepatocytes, preferably periportal hepatocytes.

Now, we know the exact genetic alteration that caused the urea cycle defect and where we need to edit to fix it. What do we fix? There are two alterations (E714X or Q335X) and fixing either one would suffice given we only need 15% restoration of function. We have adenine base editors (ABEs) and cytosine base editors (CBEs) which turn A —> G and C —> T, respectively. Consider that the E714X change occurred because a G flipped to a T. So, to fix it we need to switch a T to a G, which we cannot do with current base editors (prime editing could make this change, but that was not the chosen technology3). The opposite strand of the E714X mutation experienced a C —> A which we need to restore to a C, which we also cannot do. That leaves us with fixing the Q335X change (C —> T, opposite strand G —> A). With an ABE, we can flip the A —> G and restore the maternal allele.
Asimov Press wrote up a good explainer of genome editors and below is their image describing base editing. The NEJM paper used an ABE so mentally swap the cytosine deaminase for an adenine deaminase.
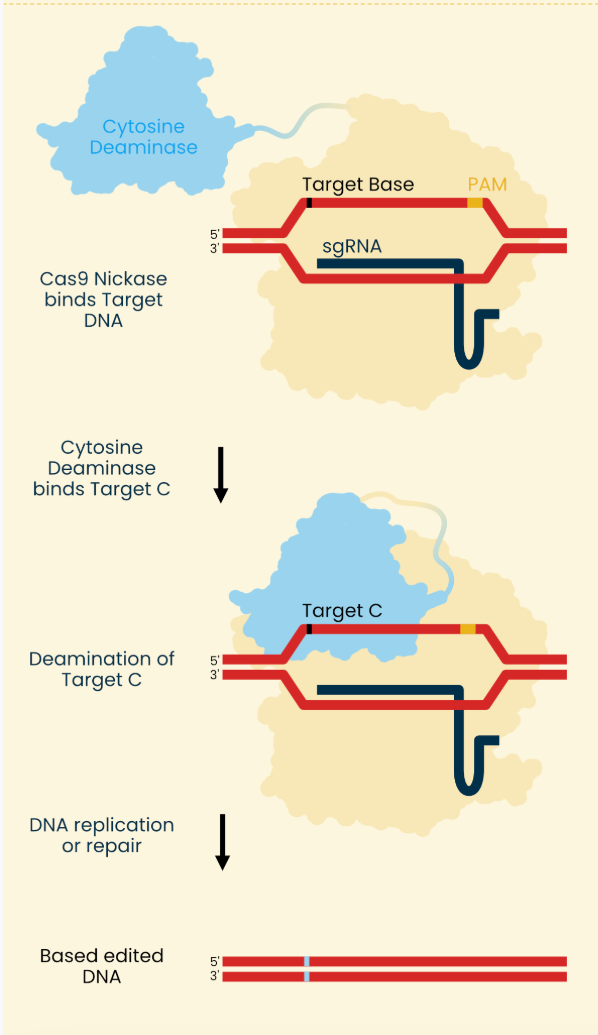
Long story short, the process can be boiled down to:
1) Two mRNA molecules enter the cell. One molecule encode cas9 and adenine deaminase and the other is the synthetic guide RNA
2) Cas9 enzyme binds the protospacer adjacent motif (PAM) found on the DNA strand
3) guide RNA binds complimentary region
The specificity of edits are tied to steps 2 and 3. The 3D structure of the region around the PAM matters in cas9 binding. For example, if the genomic region is tightly condensed or winding of the genome creates sterically unfavorable binding chemistry the cas9 will not bind well.
4) Adenine deaminase acts on exposed strand (created by binding described in 2-3 unwinding DNA at target site) turning adenine to inosine (which acts like a guanine).
5) Cas9 nickase breaks the opposite strand to what was just transformed into an inosine. This single-strand break forces the cell’s own mismatch repair process to replace this broken base with cytosine, the complimentary base to inosine.
The authors used the HuH7 cell line, a liver cancer cell line, and introduced into these cells a cassette with Q335X, E714X, and two positive controls via a lentiviral vector. A 100 base pair region centered around each variant was transduced. (The authors did not want to extract hepatocytes from KJ given the excess risk it posed to the patient, though primary hepatocytes from KJ would have been the ideal sample to use.) Now that the model system was set up, they designed seven different guide RNA’s tiling the Q335X site such that the variant base to be correct occupied different spots in the gRNA and unique distances from the PAM region.

Then used different Cas9 enzymes (SpG, SpRY) that differ primarily in their PAM preferences. SpG recognizes NGN PAM structures where N is any base, and SpRY is PAM-less. They also tested different ABEs - ABE8e [broadest edit window], ABE8.20 [intermediate] and ABE8.8 [narrowest]. The edit window is the distance from the distal end of the PAM that the ABE can chemically modify. All together, they tested different gRNA, ABE, and Cas9 combinations for the percentage of aligned sequencing reads with the edit.

SpG-AbE8e-gRNA8 had highest corrective editing efficiency for Q335X. They noticed that PAM associated with gRNA8 was NGC (see above figure of tiled gRNA’s schematic) so they performed a secondary screen with versions of Cas9 that prefer NGC PAM’s to boost specificity since SpG prefers a very generic NGN PAM. The secondary screen consisted of SpG, A1, A2, and A3 enzymes where A1-A3 each had their own set of mutations introduced (results shown below).

The left-most bar is the editing level for the reference control condition. Notice that A1-A3 offer gains in edit percentage over the base system (second from left bar). Because ABE8e has been reported to have gRNA-independent off-target editing, the ABE8e-V106W variant, which largely eliminates this off-target editing of ABE8e was used in a final transfection screen.

The A1 system had the best edit percentages when the V106W variant was introduced. Therefore, the A1-ABE8e-V106W/gRNA8 system was selected. The final drug product k-abe is a gRNA (20 nt spacer corresponding to protospacer DNA sequence matching CPS1 gene spanning Q335X + 80 nt tracrRNA domain that creates binding handle for nickase) + mRNA (encoding ABE8e-V106W-SpCas9D10A with a nuclear localization signal [NLS]) packaged in a lipid nanoparticle (LNP). The LNP design was taken from an earlier study that performed base editing of PCSK9 to lower cholesterol in primates. Acuitas therapeutics made this LNP from a mixture of the lipid components ALC-0307, PEG-lipid ALC-0159, 1,2-distearoyl-sn-glycero-3-phosphocholine, and cholesterol.
We previously established that the LNP must deliver the payload to periportal hepatocytes. The LNP is being infused intravenously and quickly binds ApoE in the bloodstream (Acuitas Therapeutics). ApoE-LNP can now bind LDL-R, CLDL-R, and ApoE-R2. ApoE binding affinity to its receptors is highest when ApoE is bound to lipid. LDL-R is expressed in different tissues including high expression in Leydig cells of the testes, adrenal gland, lung, stomach, duodenum, colon, liver, and kidney. Presumably the LNP is delivered to these tissues where it is endocytosed and subsequently protonated (intracellular vesicles are acidic) allowing for release of RNA payload into the cytosol. There is no periportal hepatocyte specific targeting agent yet the ApoE mechanism allows for adequate tissue specific targeting. Ideally, there would be less or no distribution to other organs. A well-designed LNP can ensure that the RNA product only touches the cells of interest. Recently, SORT LNPs and DUAL SORT LNPs and targeted LNPs have been developed allowing us finer control of drug distribution within the body. These are untested in trials but hold promise. We can predict protein structure well but cannot do the same for LNPs. Can we model LNP structure, tissue selectively, and intracellular protonation/payload release chemistry? Then, we can tweak lipid concentrations and component mixtures and understand their effects computationally. This could rapidly accelerate our ability to design better LNPs. End of tangent.
Before testing our drug product in animals, we need to understand k-abe’s ability to edit in an endogenous genomic context. The lentiviral cassette was randomly inserted into the HuH7 genome and thus may not reflect the editing activity in the endogenous gene locus. Reasons for edit activity differences include chromatin state differences and the cassette integrating into high transcription activity regions. To address this, they tested gRNAs that were complimentary to a region in the cassette and a region outside the cassette in the endogenous environment.

Lentiviral editing was 1.25 times greater than endogenous editing. Importantly, the rank order of gRNA performance was preserved between lentiviral and endogenous environments. Lentiviral edit activity is expected to overestimate actual edit activity by ~25% but it is a good proxy.
HuH-7 lentivirus-transduced Q335X cells tested with a clinical batch of k-abe demonstrated a maximum proportion of desired editing outcome of 67% with an EC50 of 17 ng/mL.

They tested a toxicology batch of k-abe on Rosa26-Q335X mice4 with two mice per dose and eight liver samples per mouse. At 3 mg/kg, there was 42% corrective editing of Q335X, 31% at 1 mg/kg, and 4% at 0.1 mg/kg (shown below). All observed bystander editing resulted in synonymous variants. The mice were treated at 1 month of age and were evaluated on necropsy several days later. In F1 CPS1-Q335X knock-in mice (endogenous edit of KJ variant then crossed with normal wild-type mouse to ensure 1 wild type and 1 mutant allele at CPS1 Q335), modestly higher editing rates were observed (52% at 3 mg/kg and 35% at 1 mg/kg) in line with the difference between lentiviral and endogenous cell line edits.

We know that k-abe makes the edit we want, but what others edits does it make? The BE-Hive machine learning model predicted only synonymous bystander edits that occurred within 5 base pairs of the target site. These predictions held true for the edit patterns seen in Huh-7 cells (only two non-synonymous edits totally 2.7% of reads sequenced).

Sequenced results from Q335X CPS1 deficient mice treated with k-abe revealed the same pattern of edits. In fact, the distribution of reads for each variant edit was exactly the same as the cell line. I am surprised that the distribution is exactly the same. See line by line exact number of reads for each variant in cell line work in Fig. S8 (above) and mouse work in Fig. S10 (below). Could the figure have been accidentally duplicated or perhaps I’m interpreting it wrong? Or maybe the mouse read distribution of variants is exactly that of cell lines. Should we not expect some variability on the number of reads assigned to each variant?

Let us pause here for a second to comment on an appropriate dose for baby KJ. The mouse studies showed more than 10% whole liver editing for 1 and 3 mg/kg implying that both could clinically resolve our CPS1 deficiency issue given the prior literature. Also, the authors note that 1) the LNP system was not designed for mice or cells but for primates, and 2) CPS1 is expressed in periportal hepatocytes preferentially so whole liver metrics may under-represent true edit efficacy. So, the authors chose to let safety guide the dose. More on this in a bit, but first we will consider cellular safety or off-target effects of our drug.
k-abe can, of course, make edits anywhere in the genome where the gRNA and cas9-abe complex find it chemically favorable. And, we know the LNP is going to deliver k-abe to many different tissue types. How do we assess off-target effects of k-abe? Assessing off-target effects is a regulatory requirement but the extent to which one must do it is not established. One CAR T product assessed only 20 off-target sites whereas others have evaluated thousands of sites. A summary of the k-abe off-target workflow is shown below.
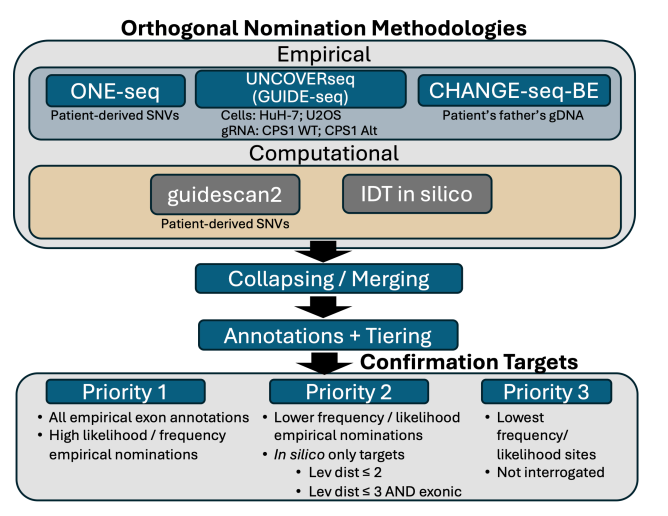
What are each of these methods?5
CHANGE-Seq: Take purified genomic DNA (in this case from KJ’s father because they could not draw enough genomic DNA from patient), use base editor, do some chemistry to tag any edit, sequence and align to identify mutated sites. Highly sensitive method for identifying mutated sites.
ONE-Seq: Synthesize short DNA strands (~300 bp) on beads including common SNPs and incubated with base editor and sequence.
UNCOVER-Seq: Add selected cas9 (without selected ABE) with gRNA and dsODN tags to HuH7 cells (primary hepatocyte preferred but did not want to biopsy patient due to risks). The tag integrates into each break caused by the cas9. We extract and purify genomic DNA and sequence tagged reads. After aligning to genome, we have identified off-target candidate sites.
Guidescan2 (pure computational): Collect each 20-mer that matches gRNA with some limit flexibility and heuristics.
IDT in silico assessment: some proprietary method that “leverages computational methods to nominate potential off-target regions for CRISPR edits”
Each method has its tradeoffs. Both CHANGE-Seq and ONE-Seq ignores chromatin context and native repair mechanisms. UNCOVER-Seq assesses the changes within cells and therefore is the most “natural”. Candidate off-target sites from each method were stacked together. For sites with at least 1-bp overlap, sites were merged and the candidate with lowest Levenshtein distance (number of edits necessary to turn candidate sequence into gRNA) was kept. Sites were prioritized as priority 1, 2, or 3 based on high frequency score from their original method, reproducibility across methods, exonic annotations. High priority sites (found in exon, higher score than on-target site) were evaluated by targeted PCR following next-generation sequencing after a clinical batch of k-abe was tested on Q335X lentivirus-transduced HuH-7 cells and primary human hepatocytes from three healthy patients (6, 7, and 13 months old). From HuH-7 cells, the estimated EC90 was 52 ng/mL and a 20x greater dose was used in the HuH-7 cells to stress test off-target effects. Of the sequenced sites, only an intronic site 369 bases upstream of closest coding sequence (a splice acceptor) at ATP7B displayed off-target editing.

ATP7B loss-of-function (LoF) causes Wilson disease but it is unlikely this finding could lead to LoF for two reasons: 1) low level editing so if edit leads to LoF it affects few cells, not enough to globally affect enough hepatocytes to mess with copper transport (see below figure), 2) unlikely that the edit itself leads to LoF given 369 bases from a splice acceptor which is the nearest coding sequence.

Primary hepatocytes from the three donors were treated at 20x the EC90 dose and sequenced three days later6. Only one site in primary hepatocytes, the ATP7B intronic site observed in HuH7 cells, displayed “even minimal off-target editing albeit at negligible levels that would not have been interpreted a priori as being indicative of off-target editing”.
Now that cellular safety has been characterized, let us move to clinical measurements of safety. AmplifyBio performed a safety study using the toxicology batch of k-abe in nonhuman primates. Nine juvenile cynomolgus monkeys (2.6-3.7 years old) with weights of 2.4 kg to 3.45 kg were split randomly while preserving mean body weights into three groups (control necropsy on day 14, 1.5 mg/kg necropsy 4 hours later, 1.5mg/kg necropsy on day 15). It’s a little unclear why 1.5 mg/kg was chosen. k-abe was infused over 30 minutes7. Lots of measurements and labs were taken including clinical observations, body weights, opthalmic observations, respiratory rates, anatomic pathology, labs (CBC, CMP, coagulation factors), organ weights, and microscopic evaluation of selected tissues.
“Administration of the drug product resulted in no adverse findings. The observed changes in the leukogram were minimal to mild and were consistent with a nonspecific physiological stress response and deemed non-adverse. The described serum enzyme elevations were minimal to mild and resolved by the conclusion of the study; therefore, these changes were considered non-adverse. The changes observed 4 hours following dose administration in the liver were transient and non-adverse as they were resolved by day 15.” From Musunuru et al 2025 NEJM Supplemental Text
The one mini-finding was increased liver weights corresponding to minimal diffuse cytoplasmic vacuolization of hepatocytes 4 hours after administration of k-abe. This was transient and resolved by day 15 hypothesized to have resulted from the LNP carrier. Toxicity from the carrier has been cited by other gene editing companies. Notably, VERVE-101 attributed the observed grade 3 transaminitis and thrombocytopenia event in their gene editing trial for lipid lowering to the LNP (note that VERVE-101’s target was LDL-R based uptake of hepatocytes). After this event, Verve shut down their trial on this asset. Subsequent trials from VERVE for lipid lowering used a different carrier and have been free of serious adverse effects. This is another reason why studying lipid chemistry, modeling lipid structure and tissue distribution is of paramount importance as we pursue more gene editing therapies.
No observable adverse events were detected at 1.5 mg/kg of k-abe in the monkeys. So, we know this to be the lower bound of a safe dose. Converting to a human equivalent dose requires scaling by body surface area. We arrive at an equivalent dose of 1.25 mg/kg. There was > 99.5% clearance of lipid excipients in the blood at 2 weeks after treatment compared to peak which informed their 3 week spacing requirement between doses to limit aggregate k-abe exposure. To be safe, the starting dose was dropped to 0.1 mg/kg.
Here is a summary of the timeline. The team was able to give KJ the first dose in ~7 months after recieving IND approval from FDA8. Incredible.

Can we shorten this timeline? What steps can we expedite? Folks like David Liu and Kiran Musunuru are the domain experts and can offer better opinions. However, here are mine. The cell line work to identify the right base editing system and mouse work to follow up whole liver editing activity data took 4 months. Can we streamline experimentation of base editing systems? I would argue the mouse work becomes less important for assessing editing activity is unnecessary if we can collect primary hepatocytes from a healthy donor (as they did for off-target work) and edit the normal genome to have the variant of interest. What about toxicology? The off target validation in mice, cell lines, and primary hepatocytes was time consuming and did not reveal anything discordant (which is good, but raises the question of what the minimal set of experiments are). Perhaps efficiency can be gained by establishing a company responsible for logistics and coordinating sub-teams executing each step of the process - a Palantir for gene editing.
Finally, lets cover KJ’s clinical course. KJ was placed on continuous renal replacement therapy (CRRT) due to his elevated ammonia shortly after birth and quickly transitioned off CRRT and onto glycerol phenylbutyrate (nitrogen scavenger), citrulline supplementation (200 mg/kg), and a protein-restricted diet. After an “expected honeymoon period from day 50-100”, KJ’s biochemical status worsened leading to an increased in the nitrogen scavenger dose to manage the elevated ammonia and glutamine and decrease in protein intake. KJ had one rotavirus-positive gastroenteritis around day 120. The first dose of the rotavirus vaccine is normally given to kids at 2 months of age. It is not stated whether KJ received his rotavirus vaccine but urea cycle disorders are not a classical contraindication to rotavirus vaccines. There were at least five distinct hyperammonemic crises (> 50 micromoles/L), and each event placed KJ at risk for permanent neurological damage and death. He was placed on the liver transplant list due to the severity of his CPS1 deficiency at 5 months.
Out of concern for KJ developing an immune response to full-length CPS1 protein (since he natively had none and his immune system might recognize it as foreign if presented on MHC class 1 proteins) prophylactic immunosupression with tacrolimus and sirolimus (exact doses not reported) was initiated on day 205. On day 208, dose 1 of k-abe (0.1 mg/kg) was given. On day 209, tacrolimus and sirolimus were re-administered. On day 230, dose 2 of k-abe (0.3 mg/kg) was given. These doses are way lower than the 1.25 mg/kg derived from the monkey experiments and the mice experiments (1 mg/kg and 3 mg/kg with max edit activity ~40%). The glycerol phenylbutyrate at 10.1 ml/m2/day kept the ammonia below 50 from day 150 onwards. And after the second dose, KJ was able to tolerate a 50% reduction in his glycerol phenylbutyrate without rising glutamate levels or other biochemical parameters. Side effects of glycerol phenylbutyrate in pediatric populations include neutropenia, fever, diarrhea, hypophagia, and cough. His first glycerol phenylbutyrate wean attempt after the first k-abe dose was unsuccessful due to rising glutamine levels.
After his first dose, KJ contracted a rhinovirus positive URI. After his second dose, he contracted gastroenteritis and a new rhinovirus or enterovirus infection. These infections may be due to long-term hospital stays, glycerol phenylbutyrate adverse effects, immunosuppression, normal (babies get sick), k-abe, or relative nutrient deficiencies. Importantly, KJ’s ammonia levels remained normal during these infections. When KJ had gastroenteritis pre-treatment, he experienced a hyperammonemic crisis. This is extraordinary. Further, KJ was able to tolerate a larger protein intake which will help him grow and develop. He remains on citrulline supplementation. k-abe allowed KJ to drop his glycerol phenylbutyrate dose by 50%, tolerate infections without hyperammonemic crises, normalize his biochemical profile, and handle a normal protein intake.

These charts suggest that the ammonia-related damage occurred from days 0-150. It remains to be seen the effect that these episodes had on KJ. Further, observing KJ as he grows will be important to understand the effects of k-abe. I am interested in following KJ’s neurocognitive testing and other parameters of brain function. Outstanding questions include KJ’s clinical and developmental history and whether he may need repeat doses of k-abe as he ages.
“Assessing the potential for unintended genomic alterations has been an established regulatory requirement in the field since the first gene editing trials in 2009. Despite such an extended track record, no universal framework for such an assessment exists in the field, except that the extent of the effort is made somewhat proportionate to the severity of the clinical picture. Thus, a CAR-T product base-edited at multiple loci advanced to 11 the clinic for treatment-refractory pediatric T-cell acute lymphoblastic leukemia (T-ALL) after 20 candidate off-target sites were evaluated, whereas some development programs for in vivo gene-editing drug products reportedly have evaluated thousands of candidate off-target sites”. We should seek to establish a standardized off-target regulatory requirement. This will help AI that FDA has implemented evaluate the content in a reproducible and accurate manner.
Link to a X thread discussing the challenges behind the variant-significance-phenotype issue once variants have been identified from WGS
I suspect the authors did not use prime editing because its 1) a more nascent technology, 2) has no FDA approvals yet so FDA may be less likely to approve their IND if it used prime editing, 3) the variant did not require prime editing, we could make do with base editing which already has FDA approval
Rosa26 is a safe-harbor locus on mouse chr6 that allows us to introduce a short cassette with the variant of interest silently. It also offers a convenient non-lethal read out option for base editing from this locus. Obviously, it is not an endogenous location and parallels the lentiviral transduction procedure for HuH7 cells.
The exact chemistry behind these methods is not included. It’s really cool and please dig into the papers but not immediately relevant for our purposes.
Primary hepatocytes had at least 4 times higher k-abe mRNA levels compared to HuH7 cells. Edit rates vary by host even amongst healthy primary hepatocytes (see below). Edit rates (both on and off-target) in primary hepatocytes should be larger than those observed in HuH7 cells. More so, it suggests that liver specific chromatin states and transcription factors favor greater edit rates. This makes sense since CPS1 and other urea cycle enzymes should be in an open chromatin state for most of the cell cycle to ensure urea can be made in the liver.

Animals received pepcid, benadryl, and dexamethasone all intramuscularly on the day prior and again up to two hours prior to treatment in order to mimic gene editing pre-treatment protocols.
An investigational new drug (IND) application is a request to FDA for permission to give an unapproved drug to humans. At a high-level INDs need pre-clinical, manufacturing controls, and clinical protocol data. There are different types of INDs of which single patient expanded access describes permissions to use for a single patient with an immediately life-threatening condition with no satisfactory alternatives or eligibility for trial enrollment in other drugs.